Researchers from the University of Michigan are using Argonne supercomputers to develop foundation models that accelerate molecular design and the discovery of new battery materials. (Image: Anoushka Bhutani, University of Michigan)
Researchers from the University of Michigan are using Argonne supercomputers to develop foundation models that accelerate molecular design and the discovery of new battery materials. (Image: Anoushka Bhutani, University of Michigan)
A University of Michigan-led team is using ALCF supercomputers to develop massive foundation models that aim to revolutionize battery materials research.
For decades, the search for better battery materials has largely been a process of trial and error.
“For most of the history of battery materials discovery, it's really been intuition that has led to new inventions,” said Venkat Viswanathan, an associate professor at the University of Michigan. “Most of the materials we use today were discovered in a relatively short window between 1975 and 1985. We’re still primarily relying on that same set of materials, with some small, incremental tweaks to improve battery performance.”
Today, advances in artificial intelligence (AI) and the computing power to support them are changing the game. With access to supercomputers at the U.S. Department of Energy’s (DOE) Argonne National Laboratory, Viswanathan and his collaborators are developing AI foundation models to speed up the discovery of new battery materials for applications ranging from personal electronics to medical devices.
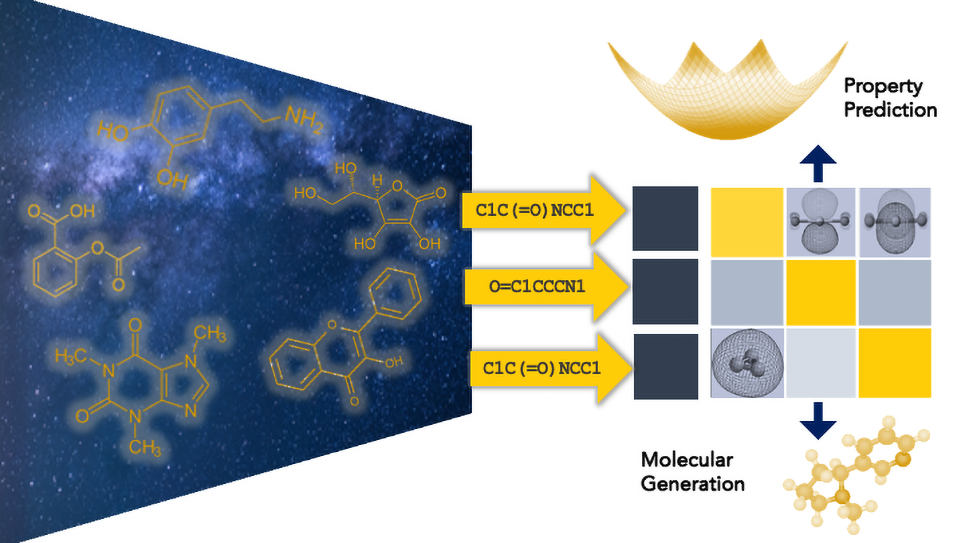
Foundation models are large AI systems trained on massive datasets to learn about specific domains. Unlike general-purpose large language models (LLMs) such as ChatGPT, scientific foundation models are tailored for specialized fields like drug discovery or neuroscience, enabling researchers to generate more precise and reliable predictions.
“The beauty of our foundation model is that it has built a broad understanding of the molecular universe, which makes it much more efficient when tackling specific tasks like predicting properties,” Viswanathan said. “We can predict things like conductivity, which tells you how fast you can charge the battery. We can also predict melting point, boiling point, flammability, and all kinds of other properties that are useful for battery design.”
The team's models are focused on identifying materials for two key battery components: electrolytes, which carry electrical charge, and electrodes, which store and release energy. Advances in both are needed to design more powerful, longer-lasting, and safer next-generation batteries.
The challenge is the scale of the chemical space for potential battery materials. Scientists estimate there could be 1060 possible molecular compounds. A foundation model trained on data from billions of known molecules can help researchers explore this space more efficiently. By learning patterns that can predict the properties of new, untested molecules, the model can zero in on high-potential candidates.
In 2024, Viswanathan’s team, including PhD students Anoushka Bhutani and Alexius Waddle, used the Polaris supercomputer at the Argonne Leadership Computing Facility (ALCF) to train one of the largest chemical foundation models to date. The model is focused on small molecules that are key to designing battery electrolytes. The ALCF is a DOE Office of Science user facility that is available to researchers from across the world.
To teach the model how to understand molecular structures, the team employed SMILES, a widely used system that provides text-based representations of molecules. They also developed a new tool called SMIRK to improve how the model processes these structures, enabling it to learn from billions of molecules with greater precision and consistency.

Available to researchers across the nation, Argonne's Aurora system provides advanced AI, simulation and data analysis capabilities to accelerate scientific discoveries and innovation. (Image by Argonne National Laboratory)
Building on this success, the researchers are now using the ALCF’s new Aurora exascale system to develop a second foundation model for molecular crystals, which serve as the building blocks of battery electrodes.
Once trained, the foundation models are validated by comparing their predictions with experimental data to ensure accuracy. This step is critical for building confidence in the model’s ability to predict a wide range of chemical and physical properties.
Prior to their work on the foundation models, Viswanathan’s team had been developing smaller, separate AI models for each property of interest. The foundation model trained on Polaris not only unified these capabilities under one roof, it also outperformed the single-property prediction models they created over the past few years.
The team is actively exploring the model’s capabilities and intends to make it available to the broader research community in the future. The team also plans to collaborate with laboratory scientists at the University of Michigan to synthesize and test the most promising candidates identified by the AI models.
Training a foundation model on data from billions of molecules requires computing power that is beyond the in-house capabilities of most research labs.
Before gaining access to ALCF supercomputers through DOE’s INCITE (Innovative and Novel Computational Impact on Theory and Experiment) program, the team was running into scaling issues. Bharath Ramsundar, a member of the INCITE project, had built AI models trained on tens of millions of molecules but found they could not match the performance of existing state-of-the-art AI models.
“There were sharp limitations in the number of molecules we could look at when training these AI systems,” said Ramsundar, founder and CEO of Deep Forest Sciences, a startup company specializing in AI-driven scientific discovery. “We started with models trained on only one million to 10 million molecules. Eventually, we reached 100 million, but it still wasn’t enough.”
The company has explored using public cloud services for some of its other research projects.
“Cloud services are very expensive,” Ramsundar said. “We’ve found that training something on the scale of a large foundation model can easily cost hundreds of thousands of dollars on the public cloud. Access to DOE supercomputing resources makes this type of research dramatically more accessible to researchers in industry and academia. Not all of us have access to the big Google-scale supercomputers.”
Equipped with thousands of graphics processing units (GPUs) and massive memory capacities, ALCF’s supercomputers are built to handle the complex demands of AI-driven research.
“There’s a big difference between training a model on millions of molecules versus billions. It’s literally not possible on the smaller clusters that are typically available to university research groups,” Viswanathan said. “You just don’t have the number of GPUs or the memory needed to scale models to this size. That’s why you really need resources like the ALCF, with supercomputers and software stacks designed to support large-scale AI workloads.”

ALCF assistant computer scientist Murali Emani (left) works with University of Michigan researchers Anoushka Bhutani, Alexius Waddle and Amal Sebastian at the ALCF INCITE GPU Hackathon. (Image: Argonne National Laboratory)
But it’s not just the computing resources that have propelled this work. The human element has also been critical. For the past two years, Viswanathan’s team has attended the ALCF’s annual INCITE hackathon to work with Argonne computing experts to scale and optimize their workloads to run efficiently on the lab’s supercomputers.
The project has also benefitted from collaborations with scientists working to employ AI in other research fields. Argonne computational scientist Arvind Ramanathan, for example, has been leading pioneering research in using LLMs for genomics and protein design. Ramanathan, who joined Viswanathan’s INCITE team, has been instrumental in applying knowledge gained from developing AI models for biology applications to battery research.
“Everyone is learning from everyone else,” Viswanathan said. “Even though we’re focused on different problems, the innovation stack is similar. There are these pockets of science, like genomics and chemistry, where the data has a natural textual representation, which makes it a good fit for language models.”
To make the foundation model more interactive and accessible, the team has integrated it with LLM-powered chatbots like ChatGPT, a novel approach that is opening the door to new possibilities for user engagement. Students, postdocs, and collaborators can ask questions, test ideas quickly and explore new chemical formulations without needing to write code or run complex simulations.
“It’s like every graduate student gets to speak with a top electrolyte scientist every day,” Viswanathan said. “You have that capability right at your fingertips and it unlocks a whole new level of exploration.”
This capability is also shifting how researchers think about the discovery process.
“It’s fundamentally changing the way we’re thinking about these things,” Viswanathan said. “These models can creatively think and come up with new molecules that might even make expert scientists go, ‘Oh wow, that’s interesting.’ It’s an extraordinary time for AI-driven materials research.”